





In Practice Series – How To Prepare For The European Medical Devices and In Vitro Diagnostic Regulations

Our monthly “in practice series” is brought to you in partnership with Kennedys law LLP
to help ensure you have access to practical insights backed by our comprehensive, in-depth regulatory expertise.
Authored by: Sarah-Jane Dobson, Partner, thomas panter, senior Associate and Myunghoon Paik, associate Kennedys Law llp
Proposed Amendments To MDR and IVDR:
On 6 January 2023, the EU Commission published its draft legislative proposal to amend the transitional provisions in Regulation (EU) 2017/745 for medical devices (“MDR”) and Regulation (EU) 2017/746 (“IVDR”) for in vitro diagnostic medical devices, hereafter “the Regulations”.
The proposed amendments introduce an extension to the transitional periods established in the Regulations to provide medical device manufacturers more time to bring their devices into conformity with the requirements of the Regulations.
This is still a proposal and needs to go through the EU legislative procedure.
However, in order to address the urgent requirements to provide more time to certify medical devices and mitigate the risk of device shortages, it is expected that the proposed amendment will be adopted by the European Parliament and the Council through an accelerated co-decision procedure.
The Legal Issue:
The MDR has been applicable since 26 May 2021 and aims to improve the safety and performance of medical devices by introducing stricter requirements for device manufacturers, including increased oversight and more stringent clinical evaluation and testing requirements.
The MDR also reinforces the transparency and traceability of medical devices by requiring manufacturers to provide more detailed information about their products.
However, these new and more stringent requirements have created several challenges for many stakeholders, namely, device manufacturers, small and medium enterprises, healthcare professionals, patients, scientific bodies and notified bodies. Regulators have also made public the significant issues they have had in developing infrastructure to support the revised regulatory regime. Some of the main issues include:
| Issue | Impact |
|---|---|
| Increased regulatory burden and cost | The MDR requires manufacturers to provide more detailed technical documentation and clinical evaluations of their devices, which can be costly and time-consuming. This may also require manufacturers to invest in costly new testing and certification processes. |
| Changes are retrospective | The changes apply retrospectively and therefore some devices that were on the market before the MDR came into effect have been reclassified as higher-risk devices, which means they now have to go through more rigorous testing and certification processes. This risks devices being removed from the market whilst they are brought into conformity, potentially leading to shortages with a corresponding risk to patient safety. |
| Conformity assessment (“Notified”) body overload | The MDR requires more manufacturers to now involve a notified body in the certification process. However, at present, only 36 notified bodies are designated under the MDR and there are reports that they have become overwhelmed by the increased workload from the new more stringent requirements. This has led to delays in the certification process. Furthermore, certain types of medical devices may require specialised notified bodies that have specific expertise in the area of the device and may not be available in all countries, which may lead to difficulties in finding the right notified body. |
Limited availability of testing laboratories | Some manufacturers are reporting difficulties in finding testing laboratories that are accredited to perform the new more stringent testing to comply with the more stringent CE marking requirements. |
The Proposed Extension And Consequences:
- MDR
Extension of the Validity of Certificates (Amendment to Article 120 (2)-(3))
The EU Commission proposed to extend the validity of certificates issued under the previous Medical Device Directive 93/42/EEC (MDD) or the Active Implantable Medical Device Directive 90/385/EEC (AIMDD) that were valid on 26 May 2021 (the effective date MDR came into force) and have not been withdrawn by a notified body.
The length of the extension to a certificate’s validity is contingent on the risk classification of the device and the manufacturer complying with certain conditions. This is laid out in the proposed amendments:
Extension of transition period for devices (Amendment to Article 120 (3))
| Device Category | Original Transition Period | New Proposed Transition Period | Cumulative Conditions To Be Granted An Extension Under The Amendment |
|---|---|---|---|
| Higher Risk Device Class III and Class IIb implantable devices, except sutures, staples, dental fillings, dental braces, tooth crowns, screws, wedges, plates, wires, pins, clips and connectors. | 26 May 2024 | 31 December 2027 | This type of medical device is covered by a certificate or declaration of conformity (“DOC”) issued under the MDD or the AIMDD before 26 May 2021; No significant changes in the design and intended purpose of the device; This type of device must not present an unacceptable risk to the health or safety of patients; users or other persons, or to other aspects of the protection of public health; By 26 May 2024, the manufacturer must put in place a quality management system (QMS) in accordance with Article 10(9) of the MDR; By 26 May 2024, the manufacturer or its authorised representative must lodge a formal application for a conformity assessment for the ‘legacy device’, and by 26 September 2024, the notified body and the manufacturer must sign a written agreement for such conformity assessment. |
| Medium and Lower Risk Devices Class IIb and Class IIa, Class Im, Is and Ir devices | 26 May 2024 | 31 December 2028 | This type of medical device is covered by a certificate or declaration of conformity (“DOC”) issued under the MDD or the AIMDD before 26 May 2021; No significant changes in the design and intended purpose of the device; This type of device must not present an unacceptable risk to the health or safety of patients; users or other persons, or to other aspects of the protection of public health; By 26 May 2024, the manufacturer must put in place a quality management system (QMS) in accordance with Article 10(9) of the MDR; By 26 May 2024, the manufacturer or its authorised representative must lodge a formal application for a conformity assessment for the ‘legacy device’, and by 26 September 2024, the notified body and the manufacturer must sign a written agreement for such conformity assessment. |
| Class III Custom-Made Implantable Devices | 26 May 2026 | The manufacturer must have applied for a conformity assessment of a device of this type before 26 May 2024, and by 26 September 2024, the notified body and the manufacturer must sign a written agreement for such conformity assessment. |
Additional requirements (Amendment to Article 120 (2))
If a certificate issued under MDD or AIMDD expires prior to the date when this proposed amendment comes into force, the extension to the validity of the certificate will be conditional upon the manufacturer having signed a contract with a notified body for the conformity assessment at the moment of the expiry.
Alternatively, if the manufacturer failed to sign a contract with a notified body to the date when this proposed amendment comes into force, a national competent authority may grant a discretionary exemption from the applicable conformity assessment procedure in accordance with Article 59 of the MDR or require the manufacturer to carry out the conformity assessment procedure within a specific time period in accordance with Article 97 of the MDR.
Removal of the “Sell-Off” Deadline for MDR (Amendment to Article 120 (4)) and IVDR (Amendment to Article 110 (4))
For devices falling under MDR, the “sell-off” date is the end date that devices should be withdrawn from the market, even if devices are already placed on the market but have not yet reached the final user.
The proposed amendment deletes the current ‘sell-off’ date (27 May 2025) in Article 120(4) MDR. Consequently, devices placed on the market before the end of the transition period can remain available for purchase on the market without a legal time restriction.
For devices falling under IVDR, the proposed amendment deletes the current ‘sell-off’ dates (25 May 2025 to 26 May 2028) in Article 110(4) IVDR. Consequently, as explained above for the MDR, in vitro diagnostic devices can also remain available on the market without a legal time restriction.
The Consequences:
The proposed amendment, if adopted, primarily aims to allow additional time for manufacturers, businesses and regulators to transition to the Regulations requirements, and to avoid device shortages.
Furthermore, the extension of the validity of existing certificates, the transition period and the removal of the “sell-off” date will alleviate supply chain disruptions and ensure patients continue to have access to safe medical devices.
However, manufacturers must ensure their products comply with the conditions set out above in order to benefit from any extension adopted.
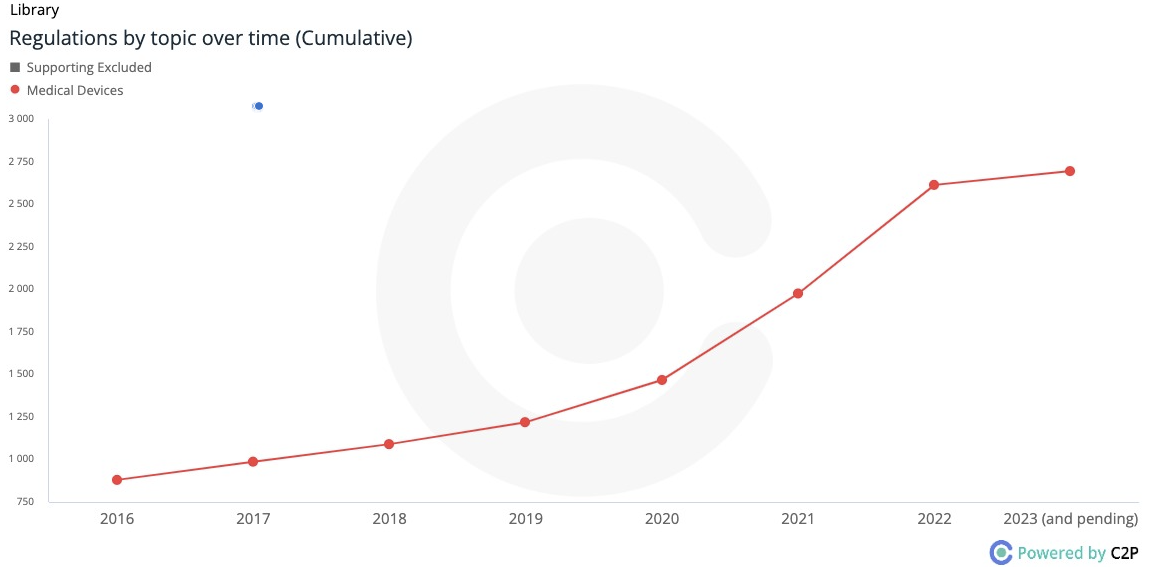
The Regulatory Trend:
C2P Regulatory Growth Chart illustrating the substantial rise in medical device regulation since 2016.
Checklist To Limit Risk:
[Downloadable version Here]
Companies should always refer to the specific regulations and guidance in place in the jurisdictions they are operating.
However, the following general principles may provide some helpful pointers for staying on the right side of the law:
- If not already, manufacturers should undertake an urgent “readiness” review of their product portfolio to include an analysis of the implications of MDR and IVDR on device categorisation, the expiry period of any existing certificates and begin contracting with notified bodies and testing houses in light of the substantially increased demand being placed on a limited resource.
- Particular attention should be given to products that may require a specialised notified body with specific technical skills, given the shortage of resources is still more acute for such devices.
- Manufacturers whose medical device certificates are due to expire should contract with a notified body as soon as possible for a conformity assessment prior to the certificate’s expiry.
In this way, if a certificate expires prior to the enforcement of the proposed amendment, the validity of such certificate can be extended. - Manufacturers whose medical device certificates have already expired or manufacturers who fail to contract with a notified body prior to the certificate expiry may apply for an exemption in accordance with Article 97 or Article 59 (1) to extend the validity of certificates.
- The extension of the validity of certificates is directly applicable without requiring notified bodies to change the date on the individual certificates.
This may pose a problem to manufacturers who export devices outside the EU as competent authorities outside the EU may not accept expired certificates.
Therefore, manufacturers need to ensure the certification process in the relevant non-EU market that may support the proposed extension of the validity of certificates in the EU. - By 26 May 2024, manufacturers must put in place a quality management system (QMS) in accordance with Article 10(9) of the MDR in order to benefit from the extended transition period.
- By 26 May 2024, the manufacturer or its authorised representative must lodge a formal application for a conformity assessment for the ‘legacy device’, and by 26 September 2024, the notified body and the manufacturer must sign a written agreement for such conformity assessment in order to benefit from the extended transition period.
- During the transitional period, manufacturers and businesses have more time to comply with the MDR and the IVDR, but it’s important to start preparing as soon as possible and to keep track of key dates and account for lead times with respect to expiry of device certificates and the end of transition periods.
- Developing and maintaining good ongoing relationships with preferred notified bodies and testing houses will ensure manufacturers are better placed to respond to future regulatory changes, particularly in times of high demand for these services.
Stay Compliant With Global Regulations
Catch up with our previous editions of our In Practice Series –
- How To Avoid “Greenwashing”,
- How To Be An ESG-Conscious Product Manufacturer,
- How To Get Ready For The New Product Liability Laws In Europe
- How To Prepare For The Upgraded Digital Services Laws In Europe
- How To Mitigate The Growing Risk From Consumer Class Actions And Collective Redress In Europe

Book Time With Our Team
Learn how C2P can help you stay ahead of regulatory changes globally and achieve uninterrupted market access.








